
If you have read any of my blogs or been in any of my lessons you will (hopefully) know how enthusiastic I am and how much I want to share my passion for Physics. Well today is no different, except I want to share my interest and hatred for a specific area of Biology (sorry Biologists)… And it’s not really Biology but instead the passing of hereditary genes.
Seeing is one of our 5 amazing senses that contribute towards a lifestyle that most of us experience and I guess to some extent… take for granted.
Why do we see?
Objects that emit light do so because of electrons dropping from a high energy level to a lower one, when this happens, the energy they had is emitted in the form of an electromagnetic wave. Different colours, different waves of light, have different energies. This is what allows us to distinguish the world around us.
When white light, which is made up of all of the colours, (like from the sun) is shone on an object, that object may absorb, reflect, refract, diffract or transmit the light. When light changes direction, or is altered in some way it means that some material, object or being is present to change the path of the light ray.
Just these two effects tells us that the world around us can be quite complex. Human beings are no different, we have organs that can process the information given to us from the world around. Take our eyes for example; they work by receiving the light, refracting it to allow us to focus and then the retina absorbs it. Each part of the eye is a marvel of creation and is of impeccable design, from the eyelid to the cornea, from the lens to the retina.
How do we see colour?
It is one thing knowing how light enters our eyes, focuses and is absorbed, but how do we see different colours? Why are some people colour blind, either by specific colours or by not seeing any at all? First of all check out the following TED talk on how our eyes identify the different colours.
Blindness
Having read or watched some of the section above, you will hopefully begin to appreciate the complexity of our eyes. A slight hiccup in cell production, too much intense light entering your eye, a bang to the side of the head, an object penetrating through the surface of the eye, these are all methods in which we could lose the sight in an eye. Unfortunately there is also hereditary conditions too which can pass down through faulty genes. Sometimes they are difficult to detect, some of them begin to manifest themselves later on in life. So a baby being born today with a faulty gene of their eye, may not show symptoms until adulthood!
Retinitis Pigmentosa
Retinitis Pigmentosa, RP is one of these faulty genes, it is thought to be hereditary and someone suffering from the conditions may be completely oblivious to the fact that they even have it until their teenage years, or perhaps even later.
Imagine, going about your daily life starting secondary school and then suddenly something begins to happen to your eye. It slowly gets progressively worse, you want to focus on your GCSEs or A Level perhaps but your eye condition keeps deteriorating…
What is RP?
The retina has two types of cells that absorb light: rods and cones. The rods are around the outer ring of the retina and are active in dim light. Most forms of retinitis pigmentosa affect the rods first. Night vision and the ability to see to the side, peripheral vision, are lost.
Cones are mostly in the centre of the retina. They help you see color and fine detail. When RP affects them, the central vision and the ability to see colour can get effected and can be lost.
Symptoms
Retinitis pigmentosa usually starts in childhood. But exactly when it starts and how quickly it gets worse varies from person to person. Most people with RP lose much of their sight by early adulthood. Then by age 40, they are often legally blind.

Because rods are usually affected first, the first symptom that may be experienced is that it takes longer to adjust to darkness (called “night blindness). For example, someone with RP may notice it when they walk from bright sunshine into a dimly lit theatre. They may trip over objects in the dark or may not be able to drive at night. They may lose their peripheral vision at the same time or soon after their night vision declines. The person may get “tunnel vision,” which means they can’t see things to the side without turning your head – as per the image above.
In later stages, the cones may be affected. This makes it harder for a person to do detailed work, and they may have trouble seeing colours. It’s rare, but sometimes the cones die first.
Someone with RP might find bright lights uncomfortable - a symptom a doctor may call photophobia. They may also start to see flashes of light that shimmer or blink. This is called photopsia.
Causes – A Bit of Biology
There are more than 60 different genes can cause the different types of RP. Parents can pass the ‘problem’ genes on to their children in three different ways:
Autosomal Recessive RP: Each parent has one problem copy and one normal copy of the gene that’s responsible, but they don’t have any symptoms. A child that inherits two problem copies of the gene (one from each parent) will develop this type of RP. Since two copies of the problem gene are needed, each child in the family has a 25% chance of being affected.
Autosomal Dominant RP: This type of retinitis pigmentosa requires only one copy of the problem gene to develop. A parent with that gene has a 50% chance of passing it to each child
X-linked RP: A mother who carries the problem gene can pass it down to her children. Each one of them has a 50% chance of getting it. Most women who carry the gene won’t have any symptoms. But about 1 out of every 5 will have mild symptoms. Most men who get it will have more severe cases. Fathers who have the gene can’t pass it to their children.
Diagnosis
An eye doctor (ophthalmologist) can tell a patient if they have RP. They’ll look into your eyes and do some specific tests:
- Ophthalmoscope: The doctor will put drops into the eyes to make the pupils wider to get a better at the retina. They will use a handheld tool to look into the back of the eye. If the patient has RP, there will be specific kinds of dark spots on the retina.
- Visual field test: The patient will look through a tabletop machine at a point in the centre of vision. While staring at that point, objects or lights will appear to the side. The patient then presses a button when he/she sees them, and the machine will create a map of how far to the side they can see.
- Electroretinogram: The eye doctor will put a film of gold foil or a special contact lens on the eye. Then he/she will measure how the retina responds to flashes of light.
- Genetic test: The patient could instead submit a DNA sample to find out which form of RP they have.
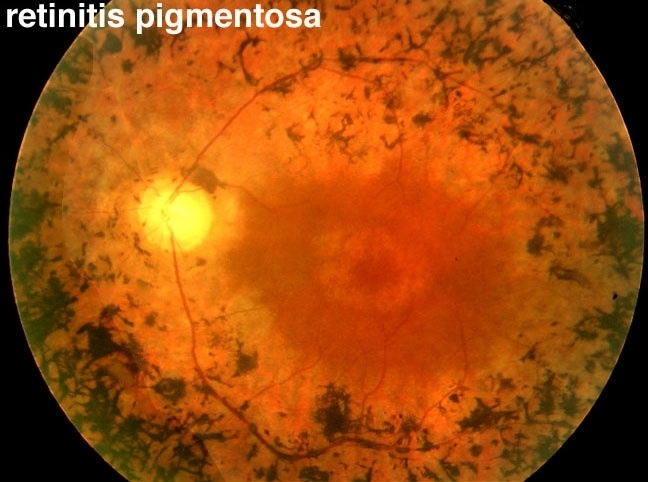
If you or anyone in your family is diagnosed with retinitis pigmentosa, all family members should go to the eye doctor for screening.
Treatment
There’s no cure for retinitis pigmentosa, but doctors are working hard to find new treatments. A few options can slow vision loss and may even restore some sight:
- Acetazolamide: In the later stages, the tiny area at the centre of the retina can swell. This is called macular edema and it too can reduce your vision. This medication can ease swelling and improve vision.
- Vitamin A palmitate: High doses of this compound may slow RP a little each year. But it has to be used with care, because too much can be toxic.
- Sunglasses: These make eyes less sensitive to light and protect your eyes from harmful ultraviolet rays that may speed vision loss.
- Retinal implant: If you have late-stage RP, a patient could be a candidate for a retinal implant that could provide partial sight. Argus II is the implant available in the US. It’s implanted into a single eye and paired with glasses equipped with a camera. Images are converted to electrical pulses that are sent to the retina. Many were able to locate lights and windows. Some were able to determine where other people were located in a room and about half of the subjects were able to read letter that were about 9 inches high.
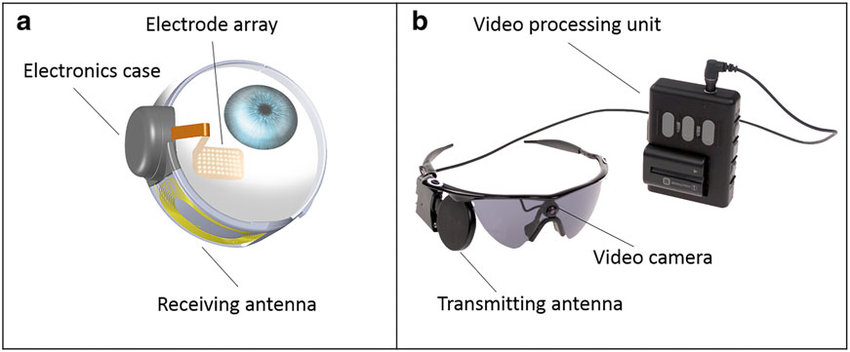
So whilst there is no cure for RP, some methods can support those people who may have the faulty genes. But imagine being able to cure it altogether? Research is constantly being done but is (like most things) limited financially. Find out more about how to help and how to support by visiting RetinaUK.org.uk – they support people affected by inherited progressive sight loss and they invest in medical research to ensure that people can lead a fulfilling life.
Want to Help?
I am trying to raise money and awareness for inherited progressive sight loss and am running the Virgin Money London Marathon on 26th April 2020. Visit my JustGiving page to find out why I am supporting this amazing charity, if your feeling generous, please support my cause.

Just by reading this page you have become more knowledgeable and understandable to those people with sight loss – thank you for taking your time read it and thank you if you have kindly donated. No matter how big or how small you donation is, it all gos to an amazing source – so thank you.